
La pandemia di COVID-19 cavalca senza sosta con 45 milioni di persone infette e quasi 1,2 milioni di morti in tutto il mondo. Ricercatori e aziende sono impegnati al massimo nello studio del virus SARS-CoV-2 per la possibile formulazione di una cura o un vaccino efficaci, una scoperta degna come minimo del premio Nobel e potenzialmente destinata a fare la storia.
È impegnata su questo fronte anche Folding@home, l’iniziativa di calcolo distribuito che sfrutta le CPU e le GPU degli utenti comuni per effettuare complesse simulazioni sulle strutture proteiche del SARS-CoV-2. La proliferazione del coronavirus ha provocato un autentico boom nelle “donazioni” di tempo-macchina da parte degli utenti, permettendo a Folding@home di raggiungere e superare i 2,4 exaFLOPS di potenza complessiva.
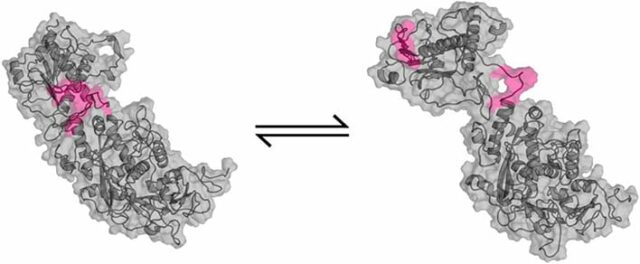
Stando a una ricerca recentemente pubblicata online, il team di Folding@home avrebbe già messo a buon frutto i miliardi di miliardi di calcoli al secondo del network distribuito scoprendo alcuni dei più inaccessibili “segreti” del SARS-CoV-2. I ricercatori hanno individuato 50 “sacche criptiche” di peplomeri, ovvero le protuberanze che permettono ai coronavirus come il SARS-CoV-2 di legarsi ai recettori della cellula ospite invadendola e dando inizio all’infezione.
Le protuberanze con i recettori chiave del coronavirus che causa il COVID-19 sono normalmente ripiegate su se stesse, ma devono prima o poi “svelarsi” per attaccare le cellule di una persona infetta. L’imponente rete di calcolo di Folding@home ha permesso di identificare, per la prima volta in assoluto, la porzione del virus necessaria allo sviluppo dell’infezione. E che ora potrà offrire ai ricercatori e alle aziende farmaceutiche un possibile bersaglio per una cura definitiva o un vaccino massimamente efficace contro la prima pandemia del 21esimo secolo.